
Smart Variant Filtering
Overview
Smart Variant Filtering (SVF) uses machine learning algorithms trained on features from the existing Genome In A Bottle (GIAB) variant-called samples (HG001-HG005) to perform variant filtering (classification).
Smart Variant Filtering increases the precision of called SNVs (removes false positives) for up to 0.2% while keeping the overall f-score higher by 0.12-0.27% than in existing solutions. Indel precision is increased by up to 7.8%, while the f-score increase is in range of 0.1 to 3.2%.
Use the Smart Variant Filtering public project
All Seven Bridges Platform users automatically have copy permissions for this project. This means you can copy the available data to your own projects on the Platform to execute analyses.
You have the options to:
- Copy the entire project - Start from the copied project and use available apps to filter a VCF file.
- Select and copy a subset of the data to your own project - Use the selected data within your own analyses in your project.
Access the Smart Variant Filtering public project
To access the Smart Variant Filtering public project:
- Click on Public projects from the top navigation bar.
- Click the title of the project, as shown below.
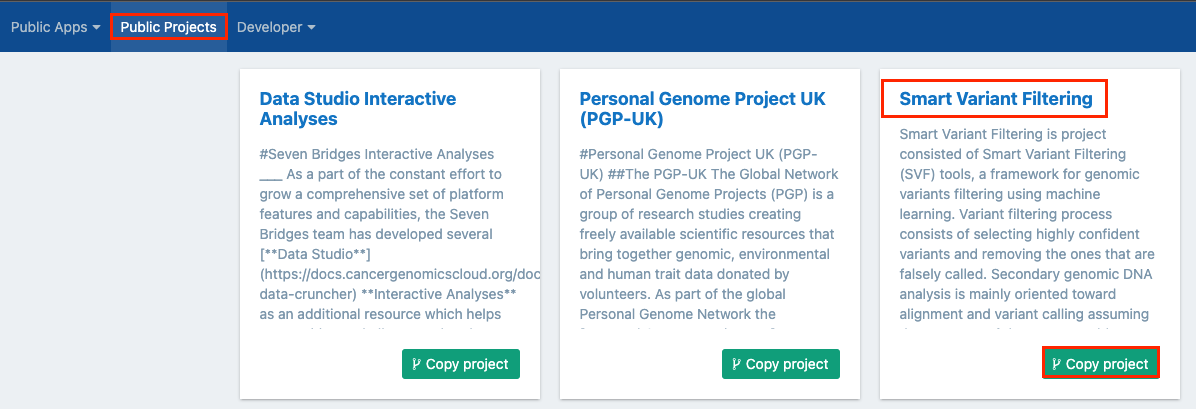
You'll be taken to the main dashboard of the Smart Variant Filtering public project. Alternatively, you can choose to copy the project, by clicking Copy project. See below for more information.
Copy the entire project
- Click Public projects in the top navigation bar.
- Locate the Smart Variant Filtering public project and click Copy project in the lower right corner.
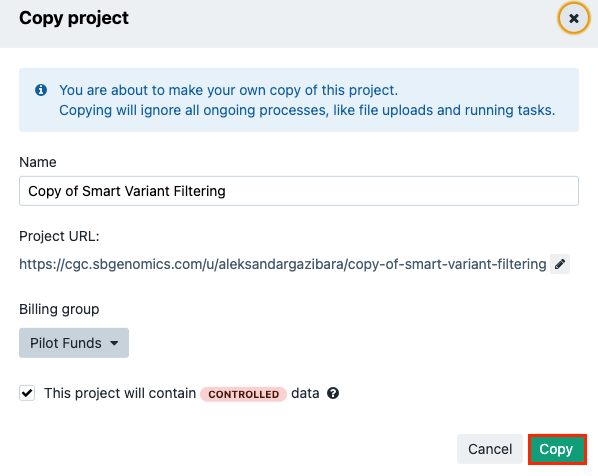
- In the pop-up window, you can name your copy of the project, select the billing group and specify whether this project will contain controlled data.
- Once you've customized the details, click Copy to copy the entire project.
You'll be redirected to the dashboard of your cloned project when it is ready, as shown below.

Learn more below on the available options once the project is copied.
Filter a VCF file
- Click the Apps tab.
- Click the run icon next to the Smart Variant Filtering tool.
- Click Select files next to a file input and choose the files in the following manner (all input files are available after copying the public project):
- Model for filtering SNVs or table to perform learning - choose
model_7_features_snv.sav. - Model for filtering indels or table to perform learning - choose
model_7_features_indel.sav. - VCF to be filtered - choose the VCF file that you want to filter.
- Click Run.
Once the task is completed, the output file, a filtered VCF file created by the tool, will be available in the Output column.
Filter large VCF files
To filter large VCF files, use the Apply Smart Variant Filtering Parallel workflow which performs filtering by parallelizing the process per chromosome. All required input files are available in your project after copying the public project.
- Click the Apps tab.
- Click the run icon next to the Apply Smart Variant Filtering Parallel workflow, which will create a draft task.
- Click Select file(s) next to a file input and choose the files in the following manner:
- dbsnp - choose
dbsnp_147.tab.vcf.gz - genome_bed_file_for_scatter - choose
human_g1k_v37_decoy.breakpoints.bed - indel_model - choose
model_6_features_indel.sav - reference - choose
human_g1k_v37_decoy.fasta - snv_model - choose
model_6_features_snv.sav - vcf - choose the VCF that you want to filter.
- Click Run.
Once the task is completed, the output file, a filtered VCF file created by the workflow, will be available in the Output column.
Train a model
To train a model that will be used for filtering a VCF, use the Smart Variant Filtering tool and provide it with tables which contain the features. All required input files are available in your project after copying the public project.
- Click the Apps tab.
- Click the run icon next to the Smart Variant Filtering tool.
- Click Select files next to a file input and choose the files in the following manner:
- Model for filtering SNVs or table to perform learning - choose
annotated_HG003_oslo_exome.tab.vcf_SNVs.table - Model for filtering indels or table to perform learning - choose
annotated_HG003_oslo_exome.tab.vcf_indels.table - VCF to be filtered - choose the VCF file that you want to filter.
- In the App Settings column, specify the machine learning algorithms:
- Machine learning algorithm for SNVs and its params - enter the classifier as well as the parameter set as comma separated values (e.g.
MLP,250,logistic,sgd) - Machine learning algorithm for Indels and its params - enter the classifier as well as the parameter set as comma separated values (e.g.
MLP,250,logistic,sgd)
- Click Run.
Once the task is completed, the output file will be available in the Output column. The result is a trained model for both SNVs and indels.
Supported classifiers and parameter sets
The currently supported classifiers and its parameters are listed in the table below.
| Classifier | Parameter set |
|---|---|
ADA | n_estimators, learning_rate,algorithm |
KNN | neighbors,algorithms,p_distance |
SVM | C,kernels |
RF | n_estimators, criterion |
QD | tol |
MLP | hidden_layer_sizes, activation,solver |
Train a model, filter variants and test the results
The entire process of training a model, applying a variant filter and benchmarking the obtained results can be done by running Smart Variant Filtering - Train, filter and test workflow. All required input files are available in your project after copying the public project.
- Click the Apps tab.
- Click the run icon next to the Smart Variant Filtering - Train, filter and test workflow.
- Click Select files next to a file input and choose the files in the following manner:
- dbsnp - choose
dbsnp_147.tab.vcf.gz - genome_bed_file - choose
genome_bed_filehuman_g1k_v37_decoy.breakpoints.bed - indel_tables - choose the following files:
annotated_ERR17432.tab.150x.vcf_indels.tableannotated_HG001-NA12878-50x.tab.vcf_indels.tableannotated_HG003.tab.hs37d5.60x.1.converted.vcf_indels.tableannotated_HG004.tab.hs37d5.60x.1.converted.vcf_indels.tableannotated_HG005.tab.150424_S1.vcf_indels.tableannotated_NA12878_CEPH_30x_ERR194147.tab.vcf_indels.tableannotated_NA12878_V2.tab.5_Robot_1_R.vcf_indels.table
- reference - choose
human_g1k_v37_decoy.fasta - region_bed_for_vcf_benchmark - choose
HG002_GRCh37_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-22_v.3.3.2_highconf_noinconsistent.bed - sdf_template - choose
1000g_v37_phase2.sdf.zip - snv_tables - choose the following files:
annotated_ERR17432.tab.150x.vcf_SNVs.tableannotated_HG001-NA12878-50x.tab.vcf_SNVs.tableannotated_HG003.tab.hs37d5.60x.1.converted.vcf_SNVs.tableannotated_HG004.tab.hs37d5.60x.1.converted.vcf_SNVs.tableannotated_HG005.tab.150424_S1.vcf_SNVs.tableannotated_NA12878_CEPH_30x_ERR194147.tab.vcf_SNVs.tableannotated_NA12878_V2.tab.5_Robot_1_R.vcf_SNVs.table
- truth_vcf - choose
HG002_GRCh37_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-22_v.3.3.2_highconf_triophased.vcf - truthset_bedfile - choose
HG002_GRCh37_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-22_v.3.3.2_highconf_noinconsistent.bed - vcf - choose
HG002-NA24385-50x.vcf
- Click Run.
The result of this task once it is completed will be trained models for SNVs and Indels, a filtered VCF and precision/recall compared to the truth set VCF.
Use a subset of the data
Instead of cloning the entire project, you can choose to select and copy a subset of the data.
- Access the public project by selecting Smart Variant Filtering from Public projectsand clicking on its title in the public projects gallery. You'll be taken to the project dashboard of the Smart Variant Filtering public project, as shown below.

- Click the Files tab in the upper lett corner. This will take you to the Files page for the Smart Variant Filtering project, as shown below.

- Filter the files or search them by:
- Keywords - You can use the search bar at the top of the page to find files by entering the file name or notes associated with a file.
- Metadata fields - Next to the search bar, you will see drop-down menus for the metadata fields Investigation, File extension, and Sample ID. Selecting a particular metadata value from one of these menus displays only files that match the value.
- You can choose specific files by selecting the corresponding checkbox in front of the file name.
- Select as many files as you desire and click Copy to.
- Select a project from the drop-down menu.
Now, you can start using the Smart Variant Filtering files you've added to your personal project in your own analysis.
Updated over 3 years ago